
技术中心
行业解决方案
随机文章选读
联系我们
- 联系人:董先生
- 联系电话:137-10902965
GMP药厂空气洁净级别
文章来源:http://www.iwuchen.com/ 2018年05月20日 点击数:6593
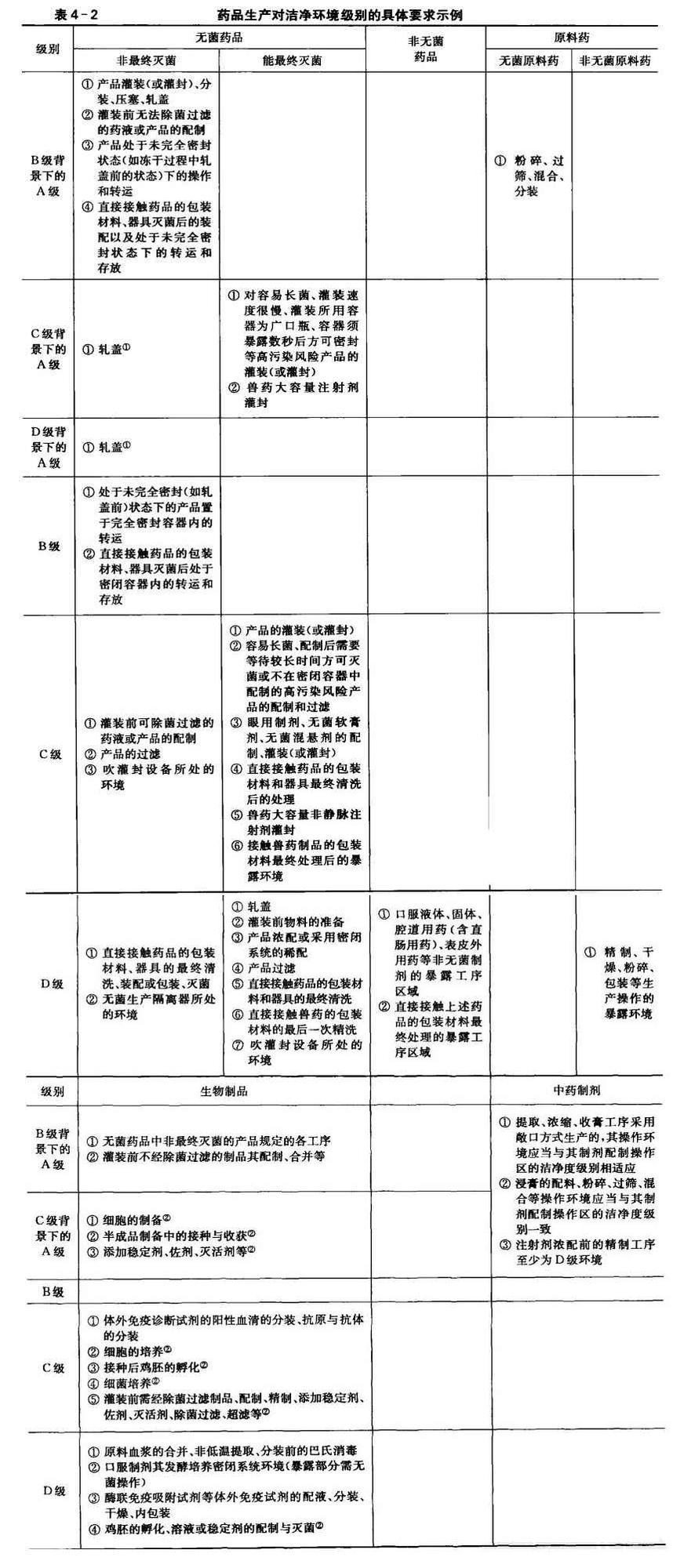 。
。