
- �����������̷���
- ��ѧ���Ӿ�������
- LED/Һ��/��·�徻������
- ʳƷ���Ͼ�������
- ��ױƷ���ջ�����������
- ҽ����е��������
- ҽԺ�����Ҿ�������
- ������ҩ��������
- �ྻʵ���Ҿ�������
- �������Դ��������
- ����/���վ�������
- ���ϻ�����������
- ���/��Ϳ��������
- �ֲ�������ϵͳ����
- �ྻ��/������վ
- ������ˮ��
- ���º�ʪϵͳ
- �ྻ�������豸����
- ���и�Ч����������
- ������/������
- ���ݴ�/�Ծ����ݴ�
- MAC/FFU������
- ��������̨
- ��Ч�����ͷ��
- ����ҩ���ྻ����������
- ��ʳƷ���Ӽ�����������ҵ�����淶���
- �����ڿ���������GB/T 18883-200���
- ��GMP���ر����°�GMP��֤�ļ������
- ���յ�����ϵͳ���й���Ҫ��
- ���ྻ���价�����������
- �����������ྻ�ҳ��������ⶨ�����
- ���ྻ�ҽྻ���������������
- �������乤��ʵʩ�й淶�����������
- ���ྻ�һ���������
- �����ͨ����ҵ����������ƶ�
- ���ྻ�Ҽ�ⱨ����ο�
- ��ҩ���ྻ������֤����
- ��ҩƷ�������������淶��¼1-�����
- ��ҽԺ����������GB 15982-2012����
- ��ϵ�ˣ�������
- ��ϵ�绰��137-10902965
��ҽ���������������淶YY 0033-2000(ȫ��������)
YY 0033-2000��ҽ���������������淶
�л�����ҽҩ��ҵ��
��ҽ���������������淶
Good manufacture practice for sterile medical devices


1 ��Χ
�����涨����ҽ���������㲿�����������������Ļ���Ҫ��
��ҽ�����߳���װ���ϵ�����ҲӦ���ϱ����Ĺ涨��
2 ���ñ�
���б������������ġ�ͨ���ڱ��������ö�����Ϊ���������ġ���������ʱ����ʾ�汾��Ϊ��Ч�����б����ᱻ����ʹ�ñ����ĸ���Ӧ̽��ʹ�����б����°汾�Ŀ����ԡ�
GB/T6583--1994 ����������������֤ ����
GB/TI6292--1996 ҽҩ��ҵ�ྻ�ң������������ӵIJ��Է���
GB/T 16293--1996 ҽҩ��ҵ�ྻ�ң��������ξ��IJ��Է���
GB/TI6294--1996ҽҩ��ҵ�ྻ�ң������������IJ��Է���
YY/T 0313--1998 ҽ�ø߷�����Ʒ��װ����־�����������
JGJ 71--1990�ྻ��ʩ�������չ淶
3 ����
����ʹ��GB/T 6583��YY/T 0313�Ķ�������ж��塣
3.1��lot
������������ȶ�ʱ�������ľ���ͬһ���ʺ����̵�ij�ֲ�Ʒȷ����������
3.2����lot number
����ʶ��������һ�����ֻ���ĸ�����֣��ݴ˿��ݺ���������Ʒ��������ʷ��
3.3������production lot
ָ��һ��ʱ���ڣ�ͬһ���������������������ľ���ͬһ���ʺ������IJ�Ʒȷ����������
ע������Щ���������IJ�Ʒ����ʱ���ѻ��������������ڹ�������Ҫ������ÿһ�����ջ����������IJ�Ʒ��Ϊ��������
3.4����� sterilization lot
��ͬһ������ڡ�ͬһ��������������ľ�����ͬ����֤ˮƽ�IJ�Ʒȷ����������
3.5���sterilization
����ʹ��Ʒ���κ���ʽ�Ĵ�������ȷ�Ϲ��Ĺ��̡�
3.6��sterile
ҽ��������������
3.7����װ primary package
����ҽ������ֱ�ӽӴ��İ�װ��
3.8��ҽ������sterile medical device
��ָ�κα����ˡ�������ҽ����е��
3.9�ྻ�ң�����clean room(area)
��Ҫ�Գ��������ﺬ�̽��п��Ƶķ��䣨�����佨���ṹ��װ���������þ����м��ٶԸ÷��䣨��������ȾԴ�Ľ��롢�����������Ĺ��ܡ�
3��10�ྻ��cleanliness
�ྻ�����ڵ�λ��������к����ڻ����ijһ�������������ӵ�����ͳ������
3.11 ��������air purification
ȥ�������е���Ⱦ���ʣ�ʹ�����ྻ����Ϊ��
3.12 ��Ա������ personnel purification room
��Ա�ڽ���ྻ�ң�����ǰ��һ��������о����ĸ������ҡ�
3.13���Ͼ�����material purification room
�����ڽ���ྻ�ң�����ǰ��һ��������о����ĸ������ҡ�
3.14����material
ָԭ�ϡ����ϣ���װ���ϡ����Э��������ȡ�
4 ������ϵ
��ҽ������������ҵӦ������ʵʩ��Ч������������ϵ���γ�һ������������������ϵ�ļ��������ڽ��й������������
ע1lGB/T 19001��YY/T 0287��GB/T 19002��YY/T 0288�涨������������ϵ��Ҫ��
4.1��������
��ӯ����Ӧ����ҵ��������������ļ�����ʽ�䲼��Ӧȷ�������ְ�ܺͲ���Ͻ�����λĿ�꼰�����������������ʵʩ��
4.2��֯����
4.2.1 ��ҵӦ��������ԥ������ϵ�Ͳ�Ʒ����Ҫ����ͨӦ����֯�������涨��ְ�ܺ����ϵ������ְ���Ȩ�ޣ����γ��ļ����Դٽ���Ч���ʹĹ�����
4.2.2����ҵ��߹�����Ӧ�ڹ�����Ա��ָ��һ������������ߴ��������涨��ְ���Ȩ�ޡ�
4.3��Ա
4.3.1 ��ҵӦ�䱸����ҽ��������������Ӧ�ľ���רҵ֪ʶ���������顢��֯��������Ϥ�����й�ҽ����е�ල�����涨�ĸ��������������Ա�ͼ�����Ա��������֯��������������������
4.3.2��ҵ��߹����߱������Ҳ�Ʒ��ӯ?��Ϥ��Ʒ��������������֯��������һ���Ŀ�ѧ�Ļ�֪ʶ���ܰ�������Ҫ����֯���������Ա�����ʵʩ�Ͳ�Ʒ������ȫ�����Ρ�
4.3.3��ҵ������ҽ���������������������������쵼��Ӧ���б�רҵ�����רҵ��ר���ϻ���֮�൱��ѧ��������ҽ����������������������ʵ�����飬���Ա�����ʵʩ�Ͳ�Ʒ��������
4.3.4��ҵ���������������������ŵĸ�����Ӧ�����뱾ְ��������Ӧ��רҵ֪ʶ�������顣����������ҽ���������������������е�ʵ������������ȷ�жϺʹ�����
��ҽ�����������������ź��������ŵĸ����˲��û�����Ρ�
4.3.5���¹ؼ���λ��������̵IJ�����Ա������������ԱӦ���и�������ѧ������רҵ������ѵ�����л�������֪ʶ��ʵ�ʲ������ܣ�רְ������Ա����Ӧ�����ʵ�������ѵ������֤�ϸڡ�
4.3��6һ������������ԱӦ�����뱾ְ��������Ӧ���Ļ��̶ȣ���רҵ������ѵ���ϸڡ�
4.3.7����ҵӦ���ձ�����Ҫ��Ը�����Ա���в�Ʒ�����������ྻ�������ƺ�ҽ������֪ʶ����Ʒ���ˡ����桢�����ȷ�������ѵ�Ϳ��ˣ����������ҵ��������������ʶ����������ѵ��¼��
4.3.8 ��Ӧ�����۲���Ӧ��������רҵ֪ʶ������ʤ����ҽ�����߹���������������Ա��
5 ������������ʩ������
5.1 ��ַ������
��ҽ������������ҵ��������������������������ĵ��桢·�漰����Ȳ�Ӧ����ҽ�����ߵ����������Ⱦ��
5.1.1 ��ַӦѡ�������������ã��������¡������������ž�Ũ�ȵ͡����к����塢��Ȼ�����õĵ�����
5.1.2��ַӦԶ����·����ͷ����������ͨҪ���Լ�ɢ����ӯ�۳����к�����Ĺ��������֡��ѳ������ؿ�����Ⱦ��ˮ����Ⱦ�����������ŵ����ྻ������������ͨ�ڵ�֮��ľ��벻��С��50 m��
5.1.3��������Ҫ��·Ӧ������·��ƽ������ѡ�����IJ��Ͻ��졣
5.1.4����Ӧ���ֺ����������������������������ö��������в���Ӱ�졣���﷿���������Ӧ����Ƨ����ȫλ�ã���Ӧ����Ӧ�İ�ȫ��ͨ������ۣ�������ʩ������ƽ���Ӧ���Ϲ����йع涨��
5.1.5������������Ӧ�ﵽ���ޣ���ˮ�����Ӳݡ�������������Ӭ�����أ���������¶���ء�
5.2��������
�����������������պͲ�Ʒ����Ҫ���Ϊһ���������ͽྻ��������Ӧ�������������̼���Ҫ��Ŀ����ྻ�ȼ�����к������֡�
5.2.1һ��������
һ��������Ӧ��ƺ������ɹ⡢ͨ�����ã��ܹ�����������Ҫ��
5.2.2�ྻ��
5.2.2.1������5.2.1���⡣��Ӧ������ྻ�ȼ�������Ӧ�Ŀ������ھ���ϵͳ����¼A��������ҽ�����߽ྻ�ң����������ྻ�ȼ��𡣸�¼B��������ҽ�����߲�Ʒ���������ྻ�ȼ��������ָ�ϡ�
5.2.2.2����ơ������װ�ྻ����ʱ��Ӧ�����������ࡣ�ྻ�ң��������ڱ���Ӧƽ�����⻬�����ѷ죬�ӿ����ܣ����������䣬��������ϴ����������ǽ�������Ľ��紦�����ɻ��λ����������ʩ���Լ��ٻҳ����ۺͱ�����ࡣ���з���������Ⱦ����ֹ������������P����������ʩ��
5.2.2.3��Ա�����ҡ��ྻ�ң��������ⴰӦ����˫�㴰�����������õ��ܷ��ԡ��ྻ�ң������Ķ��P����ྻ�ң������ڵĹܵ��������ǽ�ڻ���IJ�λ��Ӧ�ܷ⡣
5.2.2.4�ྻ�ң���������Ӧ�ܷ����ã�����ྻ�ȸߵķ�������
5.2.2.5�ྻ�ң�����Ӧ���а�ȫ�š�����ȫ��ɢ��������ƽʱ�ܷ����ã�����ʱ���ڴ���ȫͨ��Ӧ���ϰ���
5.2.2.6�ྻ�ң�������Ӧ���������̺������֣������������ֿ����̶�����
5.2.2.7�ྻ�ң������ڵ�ˮ�����������·�˰��������߹ܿڣ���װ��ǽ�ϵĸ��ֵ����豸��ǽ��ӷ촦��Ӧ�ɿ��ܷ⡣
5.2.2.8�ྻ�ң�����Ӧѡ���ⲿ���ͼ������������ڲ��õ������ƾߣ������ƾ�����װ��������������������װʱ���ƾ��붥��ӷ촦Ӧ���ÿɿ��ܷ��ʩ��
5.2.2.9����̨Ӧ�⻬��ƽ������϶�������䳾������ά����������������ϴ��������������ľ�ʻ�����̨�档
5.2.2.10�ྻ�ң�������ʹ�õ�ѹ�������������Ӧ���������������ر������Ʒʹ�ñ����ӽӴ���������ྻ��Ӧ������֤�����г�����ƣ�����Ӧ���������IJ�Ʒ��
5.2.2.11�ྻ�ң������ڵ�ˮ�ء���©���ö���ҽ�����߲�����Ⱦ��
5.3��Ա����
5.3.1��Ա������Ӧ������Ь�ҡ��������ҡ���ϴ�ң��������������ҡ���բ�һ���������ҵȡ�
5.3.2������ҽ�����������ྻ�ң��������������ྻ�ң���������ԱӦ���о�������¼D�����˽����ྻ�ң�������һ�����
5.3.3������Ա��������Ļ�Ь����ע������Ь��Ҫ������Ⱦ�����Ь�ͽ�Ҫ����ЬӦ�����Բ��������Խ�Ľ��ޡ��ڽྻ�ϣ������ڲ�Ӧ����Ь��
5.3.4��Ա����Ӧ�ϸ����شӵͽྻ��������߽ྻ��������������
5.3.5��ϴ��ˮ��ͷ����������ÿ10����1������ͷ�����˲������ֶ�ʽ��
5.3.6��բ�ҵij�����Ӧ�з�ֹͬʱ�Ĵ�ʩ�����õ��˿���������ʱ��Ӧ����������ÿ30����һ̨���ྻ�ң�����������Ա����5��ʱ������������һ��Ӧ�赥����ͨ�š�
5.3.7���ྻ�ң�����������Ա�˾����Ӧ������4 m2��
5.4���Ͼ���
5.4.1����ྻ�����������ϵ�Ӧ������ʩ���������װ�ҡ������ҵȡ�
5.4.2���Ͼ�������ྻ�ң�����֮��Ӧ������բ�һ�˫�㴫�ݴ������ڴ������Ϻ�������Ʒ��
5.4.3�������䡢��������װ��������۳�����ά�İ�װ���ϲ��ý���ྻ�ң�������ֱ�ӽӴ���Ʒ�ij���װ���������䡢����ʹ�����Ӧ����Ч��ֹ��Ⱦ�����������ܷ��װ��
5.5���ղ���
5.5.1 �ྻ������Ӧ����Ʒ�γɹ��̹����ã��������̽��ա����������ϴ���·�߾���Ҫ�̣������ڲ������̿��ƣ���������������������ϸ�ֿ�����ֹ����������
5.5.2�ྻ�ң�����ֻ�����ñ�Ҫ�Ĺ���װ������ʩ��Ӧ����������ģ����Ӧ�Ŀռ��Žྻ�ң��������������м��Ʒ���Ʒ���Ҿ����ܿ�����������ϵ��������������������еĻ�������Ⱦ�����������Ӧ���Ŵ��������ϸ����Ͳ��ϸ����������Ա�ʶ��
5.5.3�����ྻ�ȸߵĽྻ�ң������˲�������Ա���پ����������ͬ�ྻ�ȼ���Ľྻ�ң��������Ӹߵ������P�Ⲽ�á���ͬ����ྻ�ң�����֮�����ϵӦ�з�ֹ��Ⱦ��ʩ������բ�һ�˫�㴫�ݴ���
5.5.4��ͬ�����ྻ������֮������ϴ�������ô��ʹ�ʱ��Ϊ��ֹ������Ⱦ�����ʹ����˴�Խ��ǽ�����ڸ�ǽ����ֶδ��͡�
�Բ��������Ʒ�������У���ͬ�����ྻ������֮������ϴ��ݣ������ֶδ��ͣ����Ǵ���װ�ò�������������ʽ��
5.5.5���ڽྻ�ң���������ϴ�����ߣ�������ҵĿ����ྻ�ȼ���Ӧ���ƷҪ������Ӧ��100����10 000���ྻ�ң��������豸���������ڱ���������ϴ������ϴ�ҵĿ����ྻ�Ȳ�Ӧ����100 000����
5.5.6����ϴ�ӡ����P��ߴ�����������������ͨ�����õĹ��䡣��߲�Ӧ����ڽྻ�ң������ڡ�
6 �豸�빤װ
6.1�豸����ơ�ѡ��Ӧ��������Ҫ���ֺ��������ڲ�����ά�ͱ�����
6.2�ྻ�ң�������ѡ�õ��豸�빤װӦ���з���������Ⱦ��ʩ����ṹ�������͡���ת���������豸����װ��ܵ�����Ӧ��ࡢƽ���������п����������䣬������ϴ��������������ܼ�����Ⱦ��
6.3�����ϻ��Ʒֱ�ӽӴ����豸�빤װ���ܵ�����Ӧ������ʴ�������ǣ���������ϴ��������������������ϻ��Ʒ������ѧ��Ӧ��ճ����
6.4�豸���õ�������ȴ������ϴ�����ڽྻ����ͨ��ģ�߳��ͺ���ϴ����������õ���ģ���������öԲ�Ʒ�����Ⱦ��
6.5Ӧ���ö�����ģ�䣨����������ģ�ߵ�ά���ʹ�ţ��Է�ֹģ�߶Ժ����ң���������Ⱦ��
6.6Ӧ�䱸�㹻�����Ĺ�λ���ߣ������ܷ��Ժã�������ϴ���������ྻ�ң���������һ���������Ĺ�������Ҫ�ϸ�ֿ��������Ա�ǣ����ý���ʹ�á�
6.7Ӧ�����Ʊ�������ˮ���豸����ˮ������Ӧ����������Ҫ��������ˮӦ�������ڼ�⣻������ˮ�Ĵ������ܵ�Ӧ�Dz���ֻ����������ϣ�Ӧ������ϴ��������
6.8���������ͼ�����������DZ������ߡ������ȵ����÷�Χ�;���Ӧ�������������������Ҫ��Ӧ�����Ե�״̬��ʶ�������涨�����ڽ��м춨��У�顣
6.9�豸��װӦ���ڽ���ά������������֤���豸����ʱ��Ӧ������֤��ȷ���Բ�Ʒ������Ӱ��ʱ����ʹ�ã��μ���¼E��
6.10�����������豸��������Ʒ����������װ����λ���ߵĹ���Ӧ�й涨����Ӧ�����豸�����������豸ʹ�á�������ά�Ľ���¼��
7 �ɹ������Ϲ���
7.1��ҵӦ�й涨���Ʋɹ����̣����Ʋɹ��ļ���ɹ��ƻ�����ͬ������Э����ȣ���ȷ����ɹ����ϵ�����Ҫ����ȷ������ϱ��涨���������ɹ��ļ��ĸ�����
7.2Ӧ�Թ����������ۣ����������������ر����о���Ҫ��ʱ����������֤���Ƿ��з��Ϲ��ҹ涨��֤�ա������Ƚ��е��顢�������ڴ���������ǰ�Ƚ����������ã�����ϸ����������������Ӧ����ȶ���Ӧ���������湩����������¼��
7.3�ɹ����Ͻ�����Ӧ���������ԡ����顱��־�������ʼ첿�ż���ϸ���ܰ������������
7.4����Ӧ������¶ȡ����ʪ�ȷ��ϸ���Ҫ����ʴ�����塢ͨ�����á���������ʩ�IJֿ��ڣ�����Ʒ���ϸ�Ʒ�����ϸ�ƷӦ�ϸ�ֿ�����״̬��ʶ������Ч�ķ�ֹ���á��������Ϸ��ࡢ�������棬��д��λ����
7.5���Ϸ���Ӧ�м�¼���з��������˵�ǩ�������Ϸ���Ӧ��ѭ�Ƚ��ȳ���ԭ��ԭ�����ӣ�http://www.iwuchen.com/a-942/��
7.6��ǩ���ϸ�֤��ʹ��˵���顢С��װӦ��ר�˱��ܣ��䷢�š�ʹ�á�����Ӧ�м�¼��
7.7��ȼ���ױ�����Ӧ����������ʩ��
8 �ļ�
8.1������ϵ�ļ�
8.1.1��ҵӦ��������������������ϵ�������ֲᡣ
8.1.2��ҵӦ���Ʊ�����Ҫ��ij����ļ��������ļ��涨������Ч�ع᳹ʵʩ��
עl��GB/T 19001��YY/T 0287��GB/T 19002��YY/T 0288�涨�˶�����������ϵ�ļ���Ҫ��
8.2�����ļ�
��ҵӦ��ָ����Ʒ������ʹ�õļ����ļ��������ļ�Ӧͳһ����������ȷ��
8.3�ļ��Ŀ���
8.3.1��ҵӦ�����ļ����Ƴ����������йص������ļ������������ֲᡢ�����ļ����淶��ͼ�������������ļ�����ҵָ����ȣ���Ҫ���п��ơ�����ǰӦ����ʹ�õ��ļ�Ӧ����Ч�汾��Ӧ���涨�����ģ����ٱ���һ�����ϵ��ܿ��ļ����䱣������Ӧȷ������ҽ�����ߵ��������ڣ����Եõ������ߵ������淶����
8.3.2��Ϊ������¼���ļ�Ӧ�ƶ����������п��ƣ��涨������¼�ı�ʶ�����桢�����������������ںʹ��á�������¼�ı�������Ӧ������ҽ�����ߵ������������ٲ��������ꡣ
9 ��������
9.1��ҵӦ����߹�����ֱ���쵼�µ������������ţ������������ŵĸ�����Ӧ����4.3.4��Ҫ��
9.2������������Ӧ�䱸һ�����������������ͼ�����Ա����������ҽ������������ģ��Ʒ�֡�����Ҫ������Ӧ����������ѧ������ʵ���Һͼ�������豸��
9.3�����������ŵ�ְ���Ȩ�ޣ�
a)������ҽ����������ȫ���̵����������ͼ��鹤�������ݲ�Ʒ��������Ҫ���ƶ�����淶��
b)��Ȩ�����һ�����ϼ��м��Ʒ��ʹ�úͲ�Ʒ������
c)������װ���ϡ���ǩ��ʹ��˵�����Ƿ�����ʹ�ã�
d)�������ϡ��м��Ʒ����Ʒ�Ĵ��������Ƿ����ã�
e)�Բ�Ʒ���������۲죬���ڼ��飬�����۲�Ʒ�������ȶ��ԣ�ҲΪȷ����Ʒ����Ч���ṩ���ݣ�
f)��˲��ϸ�Ʒ��������;�����Ԥ����ʩ���������˻ء��ջغͲ��ϸ��Ʒ�Ĵ����취��
g)���������������豸���������Լ����������ߵĹ�����
h)����ྻ�ң������빤����ˮ�ļ��ͼ�¼��
9.4������������Ӧ���涨���н����������֤�����̼��顢��Ʒ���ռ��顣���������¼�ͱ��棻��¼�ͣ�����Ӧ��ִ�м������Ȩ��Ʒ���������ߵ�ǩ�֡�
9.5������������Ӧ���涨���г���������Ӧ�д����ԡ�
9.6������������Ӧ��ͬ�йز��ŶԹ����������ۡ�
10 �������̹���
10.1 ��ҵӦ�Բ�Ʒ�γɵ�ȫ���������̽��п��ơ�
10.2��Ʒ��ʽͶ��ǰ��Ӧ�����������յ�ȫ����֤��ȷ�����յĿ����ԡ�
10.3������̺ؼ�����Ӧ����������Ƶ㣬Ӧ�ƶ����Ƶ�����ļ�����ҵָ���ļ����繤�տ�����ҵָ����ȣ������������ļ��ӺͿ��ƣ������п��Ʋ������м�¼��
10.4���ݲ�Ʒ�����������ྻ�ȵIJ�ͬҪ���ֲ�������ֻ������ָ��������������С�����ྻ�ң���������Ա����Ʒ���밴��Ӧ��ƷҪ�����Ա�����ϵľ���������о�������ͬ�ྻ�������ڵĹ������ߵĴ��ݺ�ʹ��Ӧ��ֹ������Ⱦ��
10.5�ڽྻ�ң������ڴ�ŵ����������е��м��Ʒ��Ӧ�з�ֹ��Ⱦ�Ĵ�ʩ������Ʒ���ͼ���״̬��ʶ���Բ��ϸ���м��ƷӦ������š���¼���Է�ֹ���á�
10.6��������ϴ���������ĩ����ϴӦ����Ӧ����Ľྻ�ң�����������ӦҪ��Ĺ�����ˮ������ϴ����ϴ��ˮ����ϴ����Ӧ����ȷ�ϲ����г�����ƣ�����Ӧ�������IJ�Ʒ��
10.7��Ʒ��ʶ�Ϳ�����
10.7.1��ҽ������������ҵӦ�涨��������ȫ����ʹ�����˵ķ�����ʶ��Ʒ��ÿ����ÿ����Ʒ���γɹ�����Ӧ���������ι̵�Ψһ�Ա�ʶ�����ü�¼��ȷ�����п����ԡ�
10.7.2��ҵӦ�ƶ����ţ��������ź�������ţ������Ŀ����ļ���ÿ����ÿ����Ʒ��Ӧ�з�ӳ��Ʒʶ����Ͷ���ϡ��������̣������ྻ�ң�����������⡢�ؼ����������������������ȣ����Ŀ���������й��豸ʹ�á��������ڣ�������Ա�븴����Աǩ���ͼ�������������¼��
10.8��װ����־����ǩ��ʹ��˵����
10.8.1 ��ҽ�����߱�������ܷ��װ����װ����Ӧ���ݲ�Ʒ���ܺ��������ѡ�ĺ���ƣ���Ӧ��������Ҫ��
ע��ISO/DIS11607�涨��������ҽ��������װ���ܵĻ���Ҫ��
10.8.2��ҽ�����ߵ�����װ�����ԪӦ�ǵ���װ������װ��Ӧע�������������ͣ�����ţ�Ӧ�С���װ�����ֹʹ�á�������
10.8.3��װ�ϵı�־Ӧ����ȷָ����Ʒ�����䡢���桢�����ʹ�ã��������ԡ��������ι̣����������õ�����������������̶������ģ�����塣
10.8.4 ��װ�ϵı�־���ݱ��������Ӧ��Ʒ����Ҫ��
10.9���
10.9.1��Ҫ�������ҽ������Ӧѡ��һȷ�Ϲ����������������ø����ȷ�����Ч���Ŀɿ��ԡ�
ע�����˵����������ҽ����е������̵�ȷ���볣�����Ҫ���IS0 11131��IS011135��IS0 11137��
10.9.2���ǰ�������IJ�ƷӦ�ϸ�ֿ���Ӧ�б�ʶ���������ϸ�IJ�Ʒ�ϸ����ֿ�����
10.9.3������ԱӦ�ϸ����ļ��涨�����������������������������̼�������¼��
10.10���ϸ�Ʒ�Ŀ���
10.10.1 ��ҵӦ�ƶ����ϸ�Ʒ���Ƶij����ļ����Է�ֹ���ϸ�Ʒ�ķ�Ԥ��ʹ�û���
10.10.2�Բ��ϸ�Ʒ��Ӧ���б�ʶ���Ǽǡ����ۡ�����ʹ��á�
10.10.3���ϸ�Ʒֻ�������㷨��Ҫ�������²����ò�����;���践��Ӧȷ�Ϸ����Բ�Ʒ�IJ���Ӱ�죬������Ӧ���涨�������¼��鲢���м�¼��
10.11 ������Ԥ����ʩ
10.11.1 ��ҵӦ�ƶ���ʵʩ������Ԥ����ʩ�ij����ļ���
10.11.2��ҵӦ��Ч�����˿�Ͷ�ߺͲ��ϸ�;�������Ʒ�������̺�����������ϵ�йصIJ��ϸ������ԭ��;��ȡ������ʩ��������֤��
10.11.3��ҵӦ���ø�����Ϣ��Դ�����ֺͷ������ϸ��DZ�����أ���ȡԤ����ʩ��ʵʩ���ơ�
10.11.4��ҵ�Թ˿�Ͷ��û�в�ȡ������Ԥ����ʩ�ģ�Ӧ��¼�����ɡ�
11 ��������
��ҵӦ�ƶ����Ʒ����Ҫ���������������Ӧ�����������ļ���������ִ�С����ü�¼��
11.1 �ྻ�ң���������
11.1.1 Ӧ���ļ��涨�Խྻ�ң��������ж�����ࡢ��ϴ�����������õ��������������������ö��豸����װ�����ϺͲ�Ʒ�����Ⱦ����������Ʒ��Ӧ���ڸ�����
11.1.2Ӧ���ڰ��ո�¼C��Ҫ��Խྻ�ң��������м�⡢��¼��
11.2��������
11.2.1��ҵӦ�ƶ�������Ա������������Ӧ��������������ϴ�衢��ָ�ס�����ױ�����������Ͻ���������Ʒ���˽ྻ�ң������ȣ�����ר�˼�顣
11.2.2��ҵӦ����ְ������������ֱ�ӽӴ����ϺͲ�Ʒ�IJ�����Աÿ������Ӧ���һ�Σ��Ի��д�Ⱦ�Ժ�Ⱦ�Լ�������Ա���ô���ֱ�ӽӴ���Ʒ�Ĺ�����
11.2.3����ྻ�ң���������Ա���밴��Ӧ��ƷҪ�����Ա����������о������������ྻ������������ñ�����֡�����Ь��ֱ�ӽӴ���Ʒ�IJ�����Աÿ��һ��ʱ������ٽ���һ��������
11.3��������
11.3.1�豸���ܵ�Ӧ������ϴ���������࣬���ܡ�ð���Ρ�©����
11.3.2�豸����װ�����Ʒֱ�ӽӴ��IJ�λ������̨�桢��λ����Ӧ������ϴ�����������ֽྻ���ྻ�ң������ڵĹ�λ����Ӧ�ڽྻ�ң��������ô���ˮ������ϴ��������
11.3.3�ྻ������Ӧѡ���ʵع⻬�����������硢��������ά�Ϳ��������ʵIJ����������乤������ñ��Ӧ����Ч���ڸ����¡�ͷ������ͬ�ྻ�ȼ���ʹ�õĽྻ������Ƥ�ֱ��ڼ�������Ӧ����Ľྻ��������ϴ�����������
11.3.4 �ྻ�ң����������ڸ��������������;�������Ա���롣
12 ��Ʒ���ۺ��û�����
12.1��ҵӦ�����û��������������û���ϵ�����������û��������ʱ���û��ṩ����
12.2ÿ����Ʒ��Ӧ�����ۼ�¼��һ�����ֲ��ϸ��������أ���¼Ӧ���ٱ�������Ч�ں�һ�ꡣ
12.3���û�Ͷ���ܼ�ʱ�����������۷�����̵���ϢӦ�ܼ�ʱ�������й�ְ�ܲ��ţ���ȡ��ʩ�����м�¼��
12.4��ҵ�ɽ���ҽ�����߲����¼������ƶȣ�ָ��ר�Ż�������Ա����������Բ����¼�Ӧ��ʱ��ҩƷ�ල�������ű��档
12.5������Ʒ�¹ʱ����ƶȺͲ�Ʒ���ƶȡ���ҽ�����߳����ش���������ʱ��Ӧ��ʱ��ҩƷ�ල�������ű��档�������۲����ҳ�飬�����ѳ����IJ�Ʒ���ڲ��ϸ�Ӧ�����أ��������ϸ�Ʒ���Ƴ�����д�����
��¼A
�����ĸ�¼��
��ҽ�����߽ྻ�ң����������ྻ�ȼ����
��A1
|
|
�����������������1�� |
������������� |
||
|
�ྻ�ȼ��� |
��0.5 um |
��5 um |
�������������� |
���ξ�,��/m3 |
|
100�� |
3 500 |
0 |
l |
5 |
|
10 000�� |
350 000 |
2 000 |
3 |
100 |
|
100000�� |
3 500 000 |
20 000 |
10 |
500 |
|
300 000�� |
10500 000 |
��60 000 |
15 |
���� |
��¼B
�����ĸ�¼��
��ҽ�����߲�Ʒ���������ྻ�ȼ�������ָ��
B1 ��ҽ������Ӧ����ʹ��Ⱦ��������������������ڿ������������Ľྻ�ȼ���ʱ��Ӧ��������������������������������ܱ�֤ҽ������ʹ�ñ��治����Ⱦ������Ч�ų���Ⱦʱ�����������Ľྻ��Ӧ���������ɵ�ǰ���£�������ߡ�
B2�ྻ�ң��������ж������ʱ��Ӧ���ݸ�����IJ�ͬҪ���ò�ͬ�Ŀ����ྻ�ȼ�����������������Ҫ��������£��ྻ�ң�������������֯�ɲ��þֲ�����������������ȫ�ҿ����������ϵ���ʽ����10 000�µľֲ�100���ྻ����
B3��������ֲ�����ѭ��ѪҺ����ǻֱ�ӻ��ӽӴ�����ҽ������װ��������������㲿��������ϴ�����ļӹ���ĩ����ϴ����װ������װ�����ڵ���������Ӧ������100 000���ྻ�ȼ���ֲ�뵽Ѫ���ڵ���ҽ�����ߡ�����ij���ֲ�������ʵ��������װ�䡢��װȫ���̵���ҽ�����ߣ�Ӧ�ڲ�����10 000��������ѡ��100�����ྻ�ң�������������
B4 B3�涨�������ҽ������װ�������㲿��������ϴ�����ļӹ���ĩ����ϴ����װ������װ�����ھ�Ӧ�ڲ�����300 000���ྻ�ң������ڽ��С�
B5���Ʒ��ʹ�ñ���ֱ�ӽӴ�������ϴ��ʹ�õIJ�Ʒ����װ�������������Ľྻ�ȼ���Ӧ���Ʒ���������Ľྻ�ȼ�����ͬ����������ͬһ���������װ�����Ʒʹ�ñ���ֱ�ӽӴ������ȿ����ڲ�����300 000�ྻ�ң�������������
B6���ڲ��������������ӹ�������ֲ����ҽ�����ߣ��������ϣ���Ӧ��10 000���µľֲ�100���ྻ�ң������ڽ���������
B7�ྻ��������ϴ������ʹ��ྻ�������ҡ�ר�ù�λ���ߵ�ĩ����ϴ����������Ŀ����ྻ�ȼ���ɵ���������һ��������������������������������Ӧ��10 000���ྻ�ң������ڡ�
��¼C
�����ĸ�¼��
��ҽ�����߽ྻ�ң���������Ҫ��ա
��C1
|
|
����ָ�� |
|
|
||||
|
�����Ŀ
|
100�� |
10 000�� |
100000�� |
300000�� |
��ⷽ��
|
���Ƶ��
|
|
|
�¶�,�� |
(������Ҫ��ʱ)18-28 |
|
1���� |
||||
|
���ʪ�ȣ�% |
45��65 |
1���� |
|||||
|
���١�m��s
|
ˮƽ���� ��0.4
��ֱ���� ��0.3 |
���� |
���� |
���� |
JGJ 71--1990 |
1��/��
|
|
|
������������h |
���� |
��20 |
��15 |
��12 |
1���� |
||
|
��ѹ��,Pa |
��ͬ����ྻ�ң��������ྻ�ң����� ��ǽྻ�ң�����֮���5 |
1���� |
|||||
|
�ྻ�������������������10 |
|||||||
|
������ |
��0.5 um |
��3500 |
��350 000 |
��3500 000 |
��10 600 000 |
|
|
|
����m3 |
��5 pm |
0 |
��2 000 |
��20 000 |
��60 000 |
GB/TI6292--1996
|
1����
|
|
�����������m3 |
��5 |
lOO |
500 |
|
GB/T16293--1996 |
1���� |
|
|
���������������� |
��l |
��3 |
��10 |
��l5 |
GB/T16294--1996 |
1���� |
|
˵����
��ҽ�����߽ྻ�ң������ھ�̬�����¼��ij����������ξ������������������������100���������٣�����ѹ��¶ȡ����ʪ�ȱ�����Ϲ涨��Ӧ�����Ƶ�ζ������������ж�̬���ԡ�
��¼D
����ʾ�ĸ�¼��
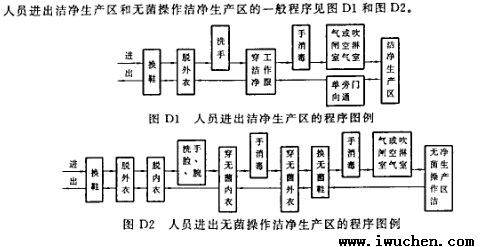
��Ա�����ྻ��������һ�����
��Ա�����ྻ���������������ྻ��������һ������ͼD1��ͼD2��
��¼E
����ʾ�ĸ�¼��
��֤��ȷ��
E1��ҽ������Ͷ������ǰӦ���ྻ��������Ҫ��ʩ���豸����װ�����ս�����֤��ȷ�ϡ�
E2 ��Ӱ���Ʒ����Ҫ���أ��繤�ա���װ���������Ʒ�������Ҫԭ���ϡ���Ҫ�����豸�����ı�ʱ���Լ�����һ�����ں�Ӧ����������֤��ȷ�ϡ�
E3Ӧ������֤��ȷ�϶��������֤��ȷ����Ŀ���ƶ���֤��ȷ�Ϸ���������֯ʵʩ��
E4��֤��ȷ�Ϲ�����ɺ�Ӧ��Ӧ�γ��ļ�����֤��ȷ���ļ�Ӧ������֤��ȷ�Ϸ�������֤��ȷ�ϱ��桢���ۺͽ��顢���˵ȡ�
E5��֤��ȷ�Ϲ������γɵ��ļ�Ӧ�鵵���档
E6��ҽ��������������֤��ȷ�ϵ���Ŀ�������������ڣ���
a)��������ϵͳ��
b)��Ҫ�����豸��
c)�ؼ��������ա���װ��������
d)��Ҫԭ�����ϱ����
e)������ˮϵͳ��ĩ����ϴ���գ�����У���
f)������ѹ���������������壨����У���
g)����豸���̣�����У���
��¼F
����ʾ�ĸ�¼��
����Ŀ¼
[1] GB/T 19001--199.1 ������ϵ ��ơ���������������װ�ͷ����������֤ģʽ
[2]GB/T 19002--1994 ������ϵ ��������װ�ͷ����������֤ģʽ
[3]YY/T0287--1996 ������ϵ ҽ����еGB/T 19001--IS0 9001Ӧ�õ�ר��Ҫ��
[4] YY/T 0288--1996������ϵ ҽ����еGB/T 19003--IS0 9002Ӧ�õ�ר��Ҫ��
[5] IS011134��1994 ҽ�Ʊ�����Ʒ���������ȷ�Ϻͳ������Ҫ����ҵʪ�����
[6] ISOl1135��1994 ҽ����е�����������������ȷ�Ϻͳ������
[7] IS0 11137��1995 ҽ�Ʊ�����Ʒ�����һһȷ�Ϻͳ������Ҫ���������
[8] ISO/DIS 11607:1999ҽ����е���������װ
������Դ��http://www.iwuchen.com/
GMP��:ҩƷ�������������淶(2010����)(���������79�ţ�
�°�GMP��:ҩƷ�������������淶��¼1-��ҩƷ
ҩƷ�������������淶��֤�����취(����2011��ȫ��)
��ҩ�������������淶��ѵָ��(��ҩGMP��ѵ�̲�)
ISO15378-2011ҩƷԭʼ��װ����-���������淶���İ�
��һ����ʳƷ�����ィ���̽���-������ʳƷ����ر���һ��������ʳƷ���������淶GB17405-1998(ȫ��������)
��������
- �� ����ױƷ�������������淶��2022��
- �� ���������������ϰ����ǽʩ�����ձ�
- �� GB50346-2011 ���ﰲȫʵ���ҽ��������淶(ȫ��)
- �� GB24461-2009 �ྻ���õƾ���Ҫ��ȫ�İ棩
- �� ���������FS209Eȫ�İ棨�ྻ�ұ���
- �� ���������������ϸ��2017�棩����������壩ȫ�����
- �� ��ISO14644-1��2015�ı仯������
- �� ��������������淶
- �� ���ý�����ůͨ�������������ƹ淶GB50736-2012(����
- �� ��ԭ����ʵ�������ﰲȫͨ����WS233-2017��ȫ�����
�Ƽ�����
- �� ����������������ʵ�������ﰲȫ�������ȫ���������
- �� ʵ�鶯�﷿��ʩ����������(SOP)д����
- �� ���ﰲȫʵ���ҵȼ����ֱ�
- �� һ���������Ͳ��������ҵ��������ʵʩ
- �� ����ʽGIS��װר�÷����ﳧ��|330KV���վGIS�������
- �� �ྻ��ʩ���ֳ���δ�����������?
- �� ��β�����������ҩ���������ӵ��ܷ�Ҫ��
- �� ��ͬ���������Ľྻ������Щ�ص㣿
- �� GIS������|500KV������|gis�����������
- �� �ɾ��ؾ��ÿ������ྻ����һ�ڹ��̣�EPC�����ܳа�����
���ڡ���ɽ����ݸ����ɽ�����š����ݡ����ݾ������̹�˾|������|�ྻ��|����|��������|��������|�ྻ����|�ྻ����|�ྻ��|GMP����|GMP�����ȿյ��ྻ����ʩ��������
��չ�Ķ���10���������|ʵ�鶯�﷿|�������|�ྻ�һ�������|�ྻ�����ҹ淶|GMP�ྻ�ȵȼ�|ʳƷ���������淶|�ྻ����ʩ��Ҫ��|��������װ�淶
Copyright © 2012-2018